GNN
Background
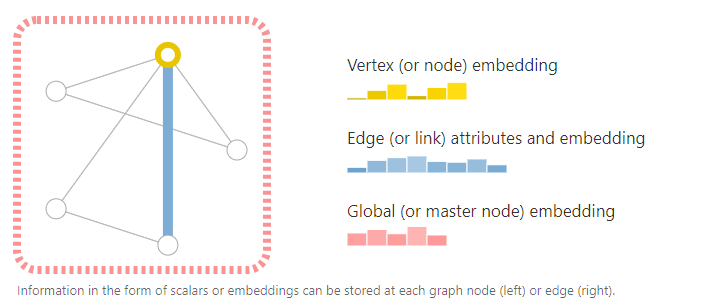
To start, let’s establish what a graph is. A graph represents the relations (edges) between a collection of entities (nodes).
To further describe each node, edge or the entire graph, we can store information in each of these pieces of the graph.
We can additionally specialize graphs by associating directionality to edges (directed, undirected).
Tasks:
- Node Classification: Classifying individual nodes.
- Graph Classification: Classifying entire graphs.
- Node Clustering: Grouping together similar nodes based on connectivity.
- Link Prediction: Predicting missing links.
- Influence Maximization: Identifying influential nodes.
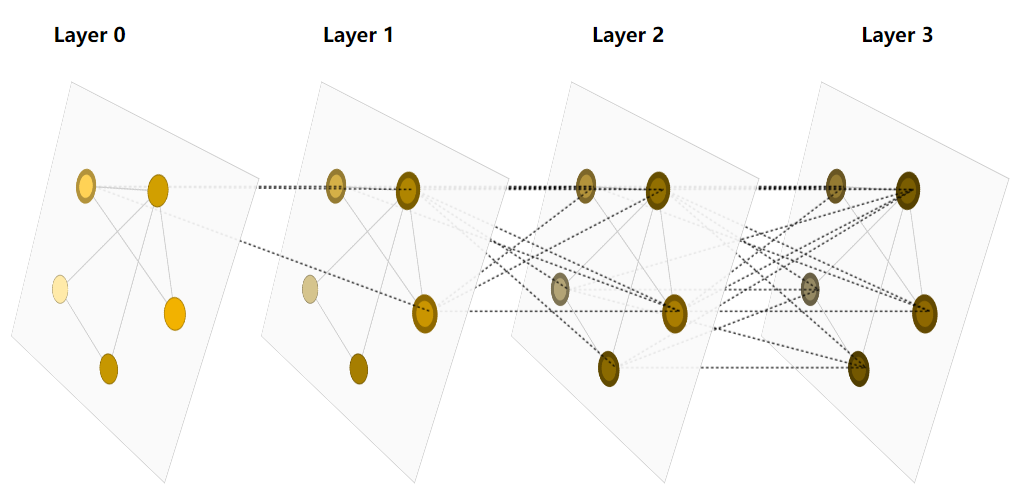
A GNN is an optimizable transformation on all attributes of the graph (nodes, edges, global-context) that preserves graph symmetries (permutation invariances).
GNNs adopt a “graph-in, graph-out” architecture meaning that these model types accept a graph as input, with information loaded into its nodes, edges and global-context, and progressively transform these embeddings, without changing the connectivity of the input graph.
The design space for our GNN has many levers that can customize the model:
- The number of GNN layers, also called the depth.
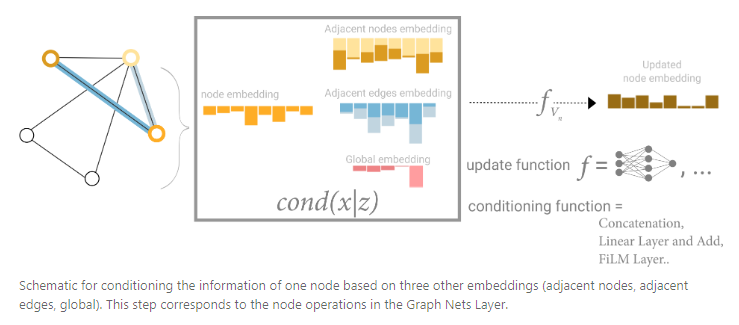
- The dimensionality of each attribute when updated. The update function is a 1-layer MLP with a relu activation function and a layer norm for normalization of activations.
- The aggregation function used in pooling: max, mean or sum.
- The graph attributes that get updated, or styles of message passing: nodes, edges and global representation.
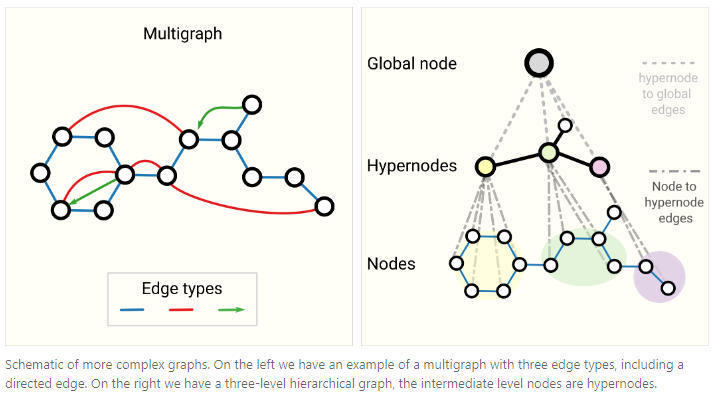
Other types of graphs
- multigraphs
- hypergraphs
- hypernodes
- hierarchical graphs
The Challenges of Computation on Graphs:
-
Lack of consistent structure, Graphs are extremely flexible mathematical models; but this means they lack consistent structure across instances. Consider the task of predicting whether a given chemical molecule is toxic, the following issues quickly become apparent:
- Molecules may have different numbers of atoms.
- The atoms in a molecule may be of different types.
- Each of these atoms may have different number of connections.
- These connections can have different strengths.
Representing graphs in a format that can be computed over is non-trivial, and the final representation chosen often depends significantly on the actual problem.
-
Node-Order Equivariance, eg. The same graph labelled in two different ways.
-
Scalability, Graphs can be really large! Think about social networks like Facebook and Twitter, which have over a billion users. Operating on data this large is not easy. Luckily, most naturally occuring graphs are sparse
Introduce operation to GNN:
- Extend convolutions to Graphs
- Polynomial Filter on Graphs, Polynomial Filters are Node-Order Equivariant
- The Graph Laplacian
- Polynomials of Laplacian
- ChebNet, ChebNet was a breakthrough in learning localized filters over graphs

Modern GNN
-
Embedding Computation, Message-passing forms the backbone of many GNN architectures today.
- Graph Convolutional Networks (GCN)
- Global conv: Spectral convolutions, which is Node-Order Equivariant
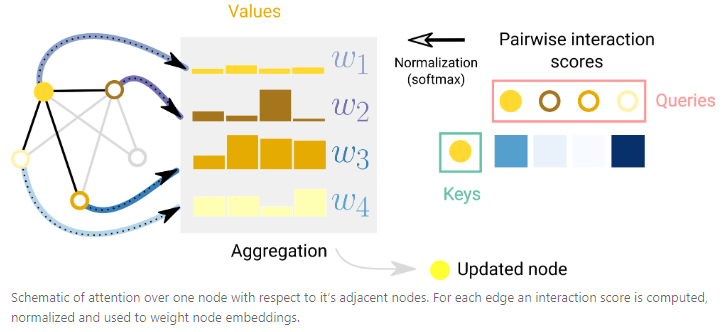
- Graph Attention Networks (GAT)
- Graph Sample and Aggregate (GraphSAGE)
- Graph Isomorphism Network (GIN)
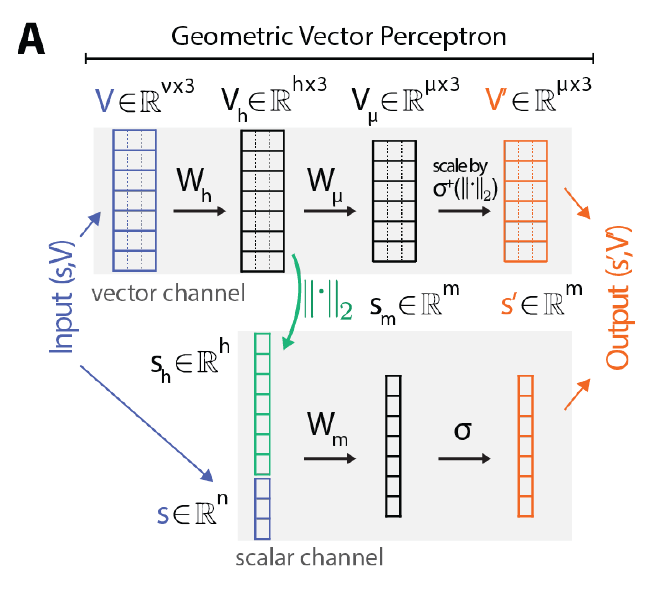
- Graph Vector Perceptions (GVP)
- Graph Convolutional Networks (GCN)
GVP-GNN
- imputed direction 插补方向
引入了一个新的模块,几何向量感知机(GVPs),以取代GNN中的密集层:
- 不需要通过节点与其所有邻居的相对方向来编码节点的方向,而只需要为每个节点表示一个绝对方向。
- 标准化了整个结构的全局坐标系,允许几何特征直接传播,而无需在局部坐标之间转换。例如,空间中任意位置的表示,包括本身不是节点的点,可以通过欧几里德向量加法轻松地在图中传播。
- 在保持标量表示法提供的旋转不变性的同时,以一种同时保留原始GNN的全部表达能力的方式执行图传播。
- 数学证明了GVP体系结构可以逼近V的任何连续旋转和反射不变标量值函数
Meta-ESM, Evolutionary Scale Modeling
ESM系列五篇文章的主线是利用蛋白质语言模型实现从蛋白序列预测蛋白质结构和功能,提出了ESM-1b、ESM-MSA-1b、ESM-1v、ESM-IF1、ESM-Fold五种基于Transformer的无监督的蛋白质语言模型。
-
ESM-1b: 第一篇《Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences 》介绍了该团队基于Transformer训练的顶尖水准(state-of-the-art)蛋白质语言模型ESM-1b,能够直接通过蛋白的氨基酸序列预测该蛋白的结构、功能等性质。
-
ESM-MSA-1b: 第二篇《MSA Transformer》在ESM-1b的基础上作出改进,将模型的输入从单一蛋白质序列改为MSA矩阵,并在Transformer中加入行、列两种轴向注意力机制,对位点分别计算第个序列和第个对齐位置的影响,充分利用二维输入的优势。
-
ESM-1v:第三篇《Language models enable zero-shot prediction of the effects of mutations on protein function 》中提出了ESM-1v模型,该模型与ESM-1b模型构架相同,只是预训练数据集改为UR90(ESM-1b预训练数据集为UR50)。ESM-1v为一种通用的蛋白质语言模型,能够实现蛋白质功能的zero-shot预测,即模型只需经过预训练即可应用于各种具体问题,对于特定蛋白质预测问题(例如针对特定蛋白家族)无需额外训练即可直接解决。本文中使蛋白质语言模型具有zero-shot预测能力的机制是,采用含有海量进化信息的蛋白数据库进行预训练。当所用数据库涵盖的序列足够多、足够多样(large and diverse),那么模型就有可能从数据库中学到横跨整个进化树的序列模式,那么该模型也就很可能会在预训练阶段学习到它将要应用的家族的序列模式,迁移应用时也就无需再额外训练。
-
ESM-IF1: 第四篇《Learning inverse folding from millions of predicted structures》
-
ESM-Fold:第五篇《Language models of protein sequences at the scale of evolution enable accurate structure prediction》
| Shorthand | esm.pretrained. | Dataset | Description |
|---|---|---|---|
| ESM-1b | esm1b_t33_650M_UR50S() | UR50, 12m seqs | SOTA general-purpose protein language model. Can be used to predict structure, function and other protein properties directly from individual sequences. Released with Rives et al. 2019 (Dec 2020 update). |
| ESM-MSA-1b | esm_msa1b_t12_100M_UR50S() | UR50 + MSA | MSA Transformer language model. Can be used to extract embeddings from an MSA. Enables SOTA inference of structure. Released with Rao et al. 2021 (ICML'21 version, June 2021). |
| ESM-1v | esm1v_t33_650M_UR90S_1() ... esm1v_t33_650M_UR90S_5() | UR90 | Language model specialized for prediction of variant effects. Enables SOTA zero-shot prediction of the functional effects of sequence variations. Same architecture as ESM-1b, but trained on UniRef90. Released with Meier et al. 2021. |
| ESM-IF1 | esm_if1_gvp4_t16_142M_UR50() | CATH + UR50 | Inverse folding model. Can be used to design sequences for given structures, or to predict functional effects of sequence variation for given structures. Enables SOTA fixed backbone sequence design. Released with Hsu et al. 2022. |
| 模型名称 | 输入数据类型 | 普适性 |
|---|---|---|
| ESM-1b | single sequence | family-specific |
| ESM-MSA-1b | MSA | few-shot |
| ESM-1v | single sequence | zero-shot |
ESM-1b
基因突变数据集的标签来自于临床观察, 一般是定性的标记int dtype(pathogenic, benign, uncertain),没有准确的score
Evaluation metric of unsupervised method:
- Remote Homology, 可以尝试
- Given a protein sequence x, with final hidden representation , we define the embedding of the sequence tobe a vector which is the average of the hidden representations across the positions in the sequence:
- We can compare the similarity of two protein sequences, and having embeddings and using a metric in the embedding space.
- We evaluate the L2 distance and the cosine distance . Additionally we evaluated the L2 distance after projecting the e vectors to the unit sphere.
- Given a protein sequence x, with final hidden representation , we define the embedding of the sequence tobe a vector which is the average of the hidden representations across the positions in the sequence:
- Mutation Effect, 无法使用,没有experimental score,无法计算
- in the protein dataset, through deep mutational scanning experiment, each amino acid sequence has a probability/score in float dtype, representing突变对蛋白质功能/活性的影响, 分数甚至可以 > 1, 表示突变有助于提高蛋白质活性
- To fine-tune the model to predict the effect of changing a single amino acid or combination of amino acids we regress the scaled mutational effect with:
- Where is the mutated amino acid at position , and is the wildtype amino acid. The sum runs over the indices of the mutated positions.
- As an evaluation metric, we report the Spearman between the model’s predictions and experimentally measured values. Each column consists of of all sequences in test dataset.
ESM-MSA-1b
Model modification:
- 输入为multiple sequence alignment,输入比BERT多一维,因为是multiple sequence alignment
- 1D sequence position embedding added to each row + position embedding to each column
- constraining attention to operate over rows and columns
- 预测的时候,除了待预测的sequence,需要输入other sequence to formulate MSA, 如果把整个测试集输入,数量太大,而且会影响predict效果,因为 test batch size远大于train batch size,因此提出了Random, diversity maximizing, diversity minimizing, HHFilter等下采样策略,使test batch size = train batch size
- 下游任务时,用到了row self-attention map + column self-attention map of all layers, do little/no supervision, 每个map训练/共享一个系数,weighted sum
ESM-1v
相似点:
- 都期望通过使用large and diverse dataset,让模型预训练是学习到足够多的序列模式,即使不通过finetune,zero-shot/unsupervised方式也能实现比SOTA/traditional method更好的performance;
- zero-shot的方式,提出了four methods of scoring the effects of mutations using the model,与我们task3 unsupervised方式类似:
- 比较wild type和mutant type sequence经过pre-trained model得到的predict probability间的差距
- ESM-1v的输入是做/未做 [token mask for mutant position] 的wt/mt序列,比较的是output probability of mutant position的log-odds distance
- 我们的输入是未做 [token mask for mutant position] 的wt/mt序列,比较的是output probability of acceptor/donor position的KL divergence
- achieving zero-shot: 采用含有海量进化信息的蛋白数据库进行预训练。当所用数据库涵盖的序列足够多、足够多样(large and diverse),那么模型就有可能从数据库中学到横跨整个进化树的序列模式,那么该模型也就很可能会在预训练阶段学习到它将要应用的家族的序列模式,迁移应用时也就无需再额外训练
- 用氨基酸的保守性衡量变异的影响力,若某位置与野生型相比其他氨基酸出现的概率很低,说明蛋白序列在该位点很保守,变异可能性很低,也就说明该位点的氨基酸可能对蛋白的结构和功能有重要影响。
- 预测序列变异对蛋白功能的影响的方法:输入含变异的序列:用[MASK]代替mutation token;模型预测将该token预测为mt/wt的概率的log-odds差值,作为socre。We introduce masks at the mutated positions and compute the score for a mutation by considering its probability relative to the wildtype amino acid
Here the sum is over the mutated positions, and the sequence input to the model is masked at every mutated position.
ParseError: KaTeX parse error: Undefined control sequence: \T at position 37: …_t=x_t^{mt}|x_{\̲T̲})-log p(x_t=_t… - 使用UR90数据库,含有98million条多样的蛋白序列,数据量和数据多样性都远远大于ESM-1b和ESM-MSA-1b训练所用的UR50数据库,因此训练出的模型迁移能力更强。
ESM-IF1
Predict protein sequence based on protein structure through auto-regressive training.从蛋白质骨架坐标(每个氨基酸三个原子C2N的中心坐标)中预测出它的蛋白质序列。
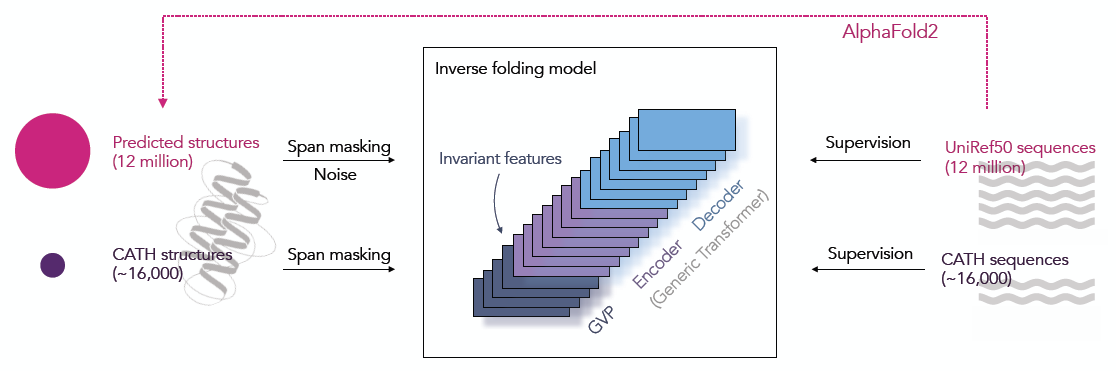
- 在已有实验性确定的蛋白质结构的基础上(16K),使用AlphaFold2预测的蛋白质结构(UniRef50)作为额外数据(12M)
- 在蛋白质骨架结构数据上达到51%的原生序列复现率,对于隐蔽残基的复现率达到72%,整体上比SOTA提升了10个百分点
- 使用Geometric Vector Perceptron(GVP)层学习向量特征的等变转换(rotation equivariant transformations of vector features)和标量特征的不变变换(rotation invariant transformations of scalar features)
- Use span masking : span masking + SBO loss (DNABert also adopt)
- zero-shot prediction evaliation metric: use the ratio between likelihoods of the mutated and wildtype sequences
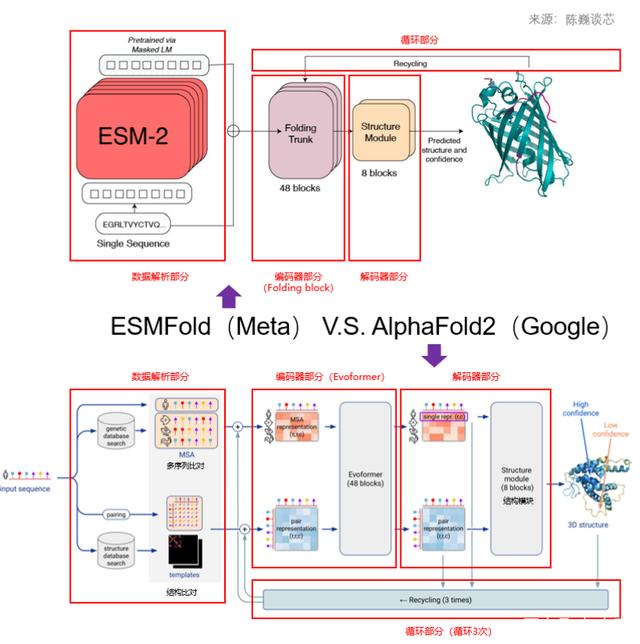
ESM-Fold
- only need single sequence as input, while AlphaFold2 need MSA. Hence achieving quick inference, 10x speed up compared with AlphaFold2
- 将AlphaFold2的MSA Folding Block模块替换为Tranformer来简化AlphaFold2中的Evoformer,加速ESMFold的关键原因
- 在AlphaFold2和RoseTTAFold中使用MSA和模板会导致两个瓶颈,首先,可能需要基于CPU检索和对齐MSA和模板,这是由于AlphaFold2和RoseTTAFold不是二维序列嵌入状态,而是使用轴向注意力对应于MSA的三维内部状态进行操作,即使使用GPU,这一计算的代价也不菲;相比之下,ESMFold是一个完全端到端的序列结构预测器,可以完全在GPU上运行,无需访问任何外部数据库。
- 虽然ESMFold速度很高,精度也不错,特别是在单序列输入的时候精度明显好于AlphaFold2;但ESMFold在多序列输入的情况下,其精度比AlphaFold2还是有差距